HISTORIA
El nombre de síndrome Proteus tiene su
origen en el mítico dios griego Proteus, también llamado el polimorfo o el viejo hombre del mar, el cual usaba sus
cambios de forma para huir de aquellos que le perseguían para usar sus
habilidades proféticas. También los pacientes afectados por este infrecuente
proceso tienen diferente aspecto en función de las zonas alteradas de su
cuerpo.
La referencia más temprana en la
bibliografía es la de Graetz en 1928 a propósito de un paciente con hipertrofia
corporal. Pocos años después, en 1979 es reconocidopor Cohen y Hayden como
entidad nosológica propia, siendo denominado síndrome Proteus por Wiedemann et
al en 1983.
Este síndrome es muy probable que haya sido
observado con anterioridad, pero con la poca fortuna de ser insuficientemente
documentado. Así, Brüning en 1904, Devousges en 1856 y Werthemann en 1952 observaron en algunos pacientes gigantismo
parcial de manos y/o pies. Se cree que «el hombre elefante» descrito a finales
del siglo XIX en Inglaterra, cuya historia fue llevada al cine en 1980 por
David Lynch, corresponde en realidad a un SP y no a una neurofibromatosis.
ETIOLOGÍA
La etiología de Síndrome Proteus es desconocida. No hay predominancia de
sexo y el análisis cromosómico es normal. Se ha propuesto una mutación somática
temprana (probablemente de los genes involucrados con los factores de
crecimiento), pues se ha encontrado una producción normal de hormona de
crecimiento (GH), pero alteración en la producción y regulación de los péptidos
que median los efectos mitógenos de la GH.
Aunque desde su primera descripción hasta la
actualidad se han descrito cerca de 150 casos, poco se sabe de su
etiopatogenia. Es un trastorno genético, esporádico, que se produce, tal vez,
por la acción de un gen letal autosómico dominante, que sobrevive por
mosaicismo somático. Dicho gen no es transmisible, porque cuando está presente
en el zigoto induce la muerte temprana del embrión.
El análisis cromosómico en estos pacientes
es normal. En un solo caso se ha detectado un segmento adicional en el brazo
largo del cromosoma 1, que sugiere una mutación somática.
Manifestaciones clínicas
Aunque ya en el momento del nacimiento se
detectan muchos de los rasgos de la enfermedad, las manifestaciones clínicas
son más evidentes en los primeros años de vida. Se han referido más de 50
rasgos patológicos diferentes, que configuran un espectro sumamente variable.
Entre todos ellos destacan:
Hemihipertrofia corporal
Aunque en ocasiones es parcial, lo habitual
es que sea completa. No existe predilección por el lado derecho o izquierdo
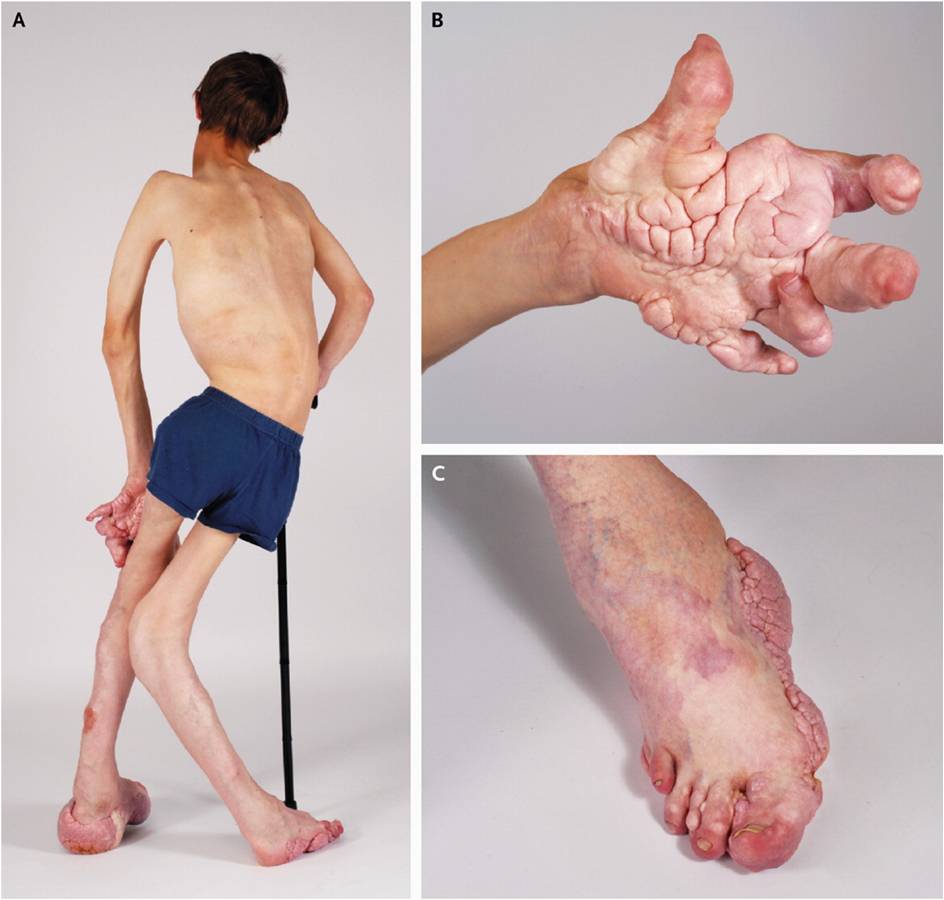
Gigantismo parcial de manos y pies
Es bilateral, aunque hay importantes
diferencias en la localización y el grado. Los metacarpos y/o metatarsos están
afectados en todos los casos. Existe normalmente una hipertrofia de los tejidos
blandos con sobre crecimiento del tejido adiposo dando, principalmente a la
superficie plantar, un aspecto cerebriforme y almohadillado.
Tumores de partes blandas
 Son prácticamente constantes y de rápido
desarrollo. Se manifiestan como lesiones voluminosas, deformantes, localizadas
en el tronco o en la parte proximal de los miembros. En general, son múltiples,
de consistencia blanda y mal delimitada. Los más frecuentes son: lipomas
hamartomas, hemangiomas cavernosos, linfangiohemangiomas, linfohemangiomas,
lipohemangiomas, hamartomas hemolinfo angiomatosos.
Son prácticamente constantes y de rápido
desarrollo. Se manifiestan como lesiones voluminosas, deformantes, localizadas
en el tronco o en la parte proximal de los miembros. En general, son múltiples,
de consistencia blanda y mal delimitada. Los más frecuentes son: lipomas
hamartomas, hemangiomas cavernosos, linfangiohemangiomas, linfohemangiomas,
lipohemangiomas, hamartomas hemolinfo angiomatosos.
Liposoma de espalda
Alteraciones óseas
Normalmente conciernen al cráneo,
observándose abultamientos, con acentuación de las protuberancias frontotemporales
de la región parietooccipital.
Crecimiento acelerado asimétrico
Suele referirse en los primeros años de
vida, aunque la talla adulta final es habitualmente normal. Los valores analíticos
de hormonas, tanto tiroideas como del crecimiento, no presentan anomalías.
Lesiones cutáneas
Las más frecuentes son de tipo pigmentario
pero pueden verse otras:
1. Nevos epidérmicos. Suelen estar presentes en más de la
mitad de los casos. A menudo presentan un patrón mixto con lesiones verrucosas
hipopigmentadas, hiperpigmentadas o ambas, que se distribuyen a lo largo de las
líneas de Blaschko.
2. Nevos melanocíticos pigmentarios. Son tan frecuentes como
los nevos epidérmicos.
3. Manchas café con leche y/o acrómicas. Se hallan presentes
en menor proporción que los nevos epidérmicos y melanocíticos pigmentarios.
4. Otras lesiones cutáneas. Entre ellas, se consideran las
siguientes: varicosidades, prominencias venosas, angioqueratomas, nevos
conectivos, neurofibromas, fibromas, disminución del tejido subcutáneo,
ausencia de mamas, etc.
DIAGNÓSTICO
Se han postulado diversos criterios
diagnósticos. Los primeros fueron propuestos por Wiedemann en 1983, en su serie inicial de 4 pacientes,
aunque ninguno de ellos presentaba la totalidad de la sintomatología descrita.
Son los siguientes: gigantismo de manos y pies,
nevos pigmentados, hemihipertrofia parcial o
completa, tumores o masas subcutáneas, anomalías craneales, crecimiento
acelerado, anormalidades viscerales.
Posteriormente, Samlaska et al, en 1989, con
una serie de 34 pacientes, y Sayama et al, en 1994, con un caso único de
síndrome Proteus, y una revisión de 63 casos, proponen una relación de
hallazgos clínicos más frecuentes. En artículos posteriores especifican que si
cuatro de los siete criterios frecuentes o mayores están presentes, el
diagnóstico de SP es seguro. El signo más específico es la presencia de una
superficie plantar de aspecto cerebriforme.
Criterios mayores
Hemihipertrofia (parcial o completa).
macrodactilia, masas subcutáneas, lipomatosis, lipomatosis pélvica, hamartomas,
hemangiomas cavernosos, linfangiohemangiomas, linfohemangioma, lipohemangioma,
hamartoma hemolinfoangiomatoso, masas plantares o palmares, nevos del tejido
conectivo, lipomatosis, exostosis craneales o en extremidades distales, nevos
epidérmicos lineales y/o espirales escoliosis
Criterios menores
Cutáneos: varicosidades, prominencias
venosas, depigmentaciones, manchas café con leche, angioqueratomas, nevos del
tejido conectivo, ausencia de mamas, neurofribroma, fibromas, disminución del
tejido subcutáneo esqueléticos: macrocefalia, crecimiento aumentado, genus valgus, cuello largo, anquilosis de codo, cifosis, pectum excavatum, retardo de crecimiento óseo, pies anchos, dislocación
de la cadera craneofaciales: estrabismo, defectos oculares, mala oclusión dentaria,
paladar arqueado, pequeña circunferencia occipitofrontal, prognatismo
mandibular, puente nasal deprimido, bajo asiento de los pabellones auriculares otros:
retaso mental, atrofia muscular, abdomen prominente, anomalías quísticas
pulmonares, crisis convulsivas, desarrollo precoz mamario, piernas musculosas,
nódulos en las cuerdas vocales, hipertrofia de pene, nariz picuda.
Algunos autores han querido ser más
estrictos dando una puntuación a cada sintomatología. Así, Hotamisligil, en
1990, otorga a la macrodactilia y/o hemihipertrofia 5 puntos, al engrosamiento
palmar o plantar 4 puntos, a los tumores subcutáneos y lipomas 4 puntos, a los
nevos epidérmicos 3 puntos, a la macrocefalia o a las exostosis craneales 2,5
puntos, y al resto de anormalidades menores 1 punto. El diagnóstico se
establece si el paciente suma 13 o más puntos. Para clarificar todos estos
criterios, en marzo de 1998 se convocó la primera conferencia nacional del SP
para padres y familiares (National Institutes of Health de Bethesda, Maryland,
EE.UU.), en donde se desarrollaron una serie de criterios diagnósticos, recomendaciones,
guías de evaluación y diagnósticos diferenciales para estos pacientes. Los
criterios diagnósticos para el SP se encuentran reflejados en la tabla IV.
Existen una serie de criterios generales que son obligatorios, como la
distribución de las lesiones en mosaico, el curso progresivo y la aparición
esporádica, y otros criterios específicos, clasificados en categorías A, B y C.
El único criterio específico incluido en la categoría A, en presencia de todos los
criterios obligatorios es suficiente para el diagnóstico, así como dos de la
categoría B o tres de la C.
Hay que destacar la descripción realizada
últimamente de otras manifestaciones de este síndrome:
1. Alteraciones oculares: exostosis periorbitarias, iris heterocromático,
coloboma retiniano, nistagmus, estrabismo, escleróticas azules, telecantus,
epibléfaron y hemimegalia del nervio óptico.
2. Alteraciones genitourinarias: quistes renales y
epididimales.
3. Manifestaciones pulmonares: tromboembolismo pulmonar y
quistes pulmonares.
4. Neuropatías: desmielinización, compresión del cuerpo
calloso, hemimegaloencefalia, nódulos calcificados subependimales, quistes
periventriculares35, craneosinostosis, epilepsia, retraso mental.
5. Otras anomalías varias: descalcificación y adelgazamiento
de la capa cortical de los huesos largos, un caso asociado a pubertad precoz,
predisposición al padecimiento de tumores y un caso con inmunodeficiencia, con
bajas cifras en sangre de IgG e IgA y linfopenia tanto de células B como T.
No obstante, debido a la amplia variabilidad
que existe dentro de este grupo de pacientes, unido a la gran similitud y
superposición que presenta este síndrome con otras enfermedades congénitas
hamartomatosas, como la neurofibromatosis, el síndrome de Kipplel-Trenaunay, la
en docromatosis, el síndrome de Mafucci y el síndrome de
Bannayan-Riley-Ruvalcaba, puede que no se esté clasificando correctamente a los
enfermos.
Evaluación del paciente con probable
síndrome Proteus.
Existen una serie de guías diagnósticas para
la evaluación de estos pacientes, que incluyen las siguientes pruebas:
– Tomografía computarizada de alta
resolución, que puede ser útil para evaluar las malformaciones quísticas
pulmonares.
– Resonancia magnética abdominal, aun en
ausencia de síntomas. Es importante para poder descubrir lipomas ocultos, que
pueden tener un comportamiento agresivo.
– Resonancia magnética del sistema nervioso
central. Imprescindible para poder descartar anomalías del sistema nervioso
central.
– Cariotipo por la posibilidad de encontrar
alguna translocación o delección.
– Radiografías óseas seriadas y evolutivas.
– Fotografías clínicas.
– Consultas a neurólogos, oftalmólogos,
hematólogosy dermatólogos, según la sintomatología y hallazgos clínicos.
IMPLICACIONES SOCIALES
En el caso del SINDROME DE PROTEUS, las
implicaciones sociales se toman desde la perspectiva de enfermedad rara la
cual se determina según el número de frecuencia que aparece. Para ser considerada como rara, cada
enfermedad específica solo puede afectar a un número limitado de la población
total, definido en Europa como menos de 1 de entre 2.000 ciudadanos (EC
Regulation on Orphan Medicinal Products). Esta cifra también se puede
expresar como 500 pacientes por cada enfermedad rara en una población de
1.000.000 de ciudadanos. Aunque 1 de 2.000, parece muy poco, en una población
total de 459 millones de ciudadanos, esto podría significar nada menos que
230.000 individuos para cada enfermedad rara.
Los
pacientes de una enfermedad rara y sus familias se ven enfrentados a la misma y
amplia gama de dificultades que surgen directamente de la rareza de estas
patologías:
• Falta de acceso al
diagnóstico correcto: el periodo entre la emergencia de los primeros
síntomas y el diagnóstico adecuado implica retrasos inaceptables y de alto
riesgo, así como diagnósticos erróneos que conducen a tratamientos inadecuados:
el laberinto del pre-diagnóstico;
• Falta de información: tanto
sobre la enfermedad misma como sobre dónde obtener ayuda, que incluye falta de
referencia a profesionales cualificados;
• Falta de conocimiento
científico: esto origina dificultades para desarrollar las herramientas
terapéuticas, para definir la estrategia terapéutica y en una palabra de los
productos terapéuticos, tanto los productos médicos como los mecanismos médicos
apropiados.
• Consecuencias sociales: El
vivir con una enfermedad rara tiene implicaciones en todas las áreas de la
vida, tanto en el colegio, elección del trabajo futuro, tiempo de ocio con los
amigos o en la vida afectiva. Puede llevar a la estigmatización (aislamiento,
exclusión de la comunidad social, discriminación para la suscripción del seguro
(seguro de vida, seguro de viaje, de hipoteca), y a menudo a oportunidades
profesionales reducidas (cuando es en absoluto relevante)).
• Falta de apropiada
calidad del cuidado de la salud: combinando las diferentes esferas de
conocimientos técnicos necesitados por los pacientes de enfermedades raras,
tales como fisioterapeuta, nutricionista, psicólogo, etc... Los pacientes
pueden vivir durante varios años en situaciones precarias sin atención médica
competente, incluyendo intervenciones de rehabilitación; permanecen excluidos
del sistema del cuidado sanitario, incluso después de haberse hecho el
diagnóstico.
Alto coste de los pocos
medicamentos existentes y cuidado:
el gasto adicional de hacer frente a la enfermedad, en términos tanto de ayudas
humanas como técnicas, combinado con la falta de beneficios sociales y
reembolsos, causa un empobrecimiento total de la familia, y aumenta
dramáticamente la desigualdad de acceso al cuidado para los pacientes de
enfermedades raras.
• Desigualdad en la
disponibilidad de tratamiento y cuidado: tratamientos innovadores están a
menudo desigualmente disponibles en Unión Europea a causa de los retrasos en la
determinación del precio y/o en la decisión de reembolso, falta de experiencia
de los médicos que tratan (no hay médicos suficientes implicados en las pruebas
clínicas de enfermedades raras) y la ausencia de recomendaciones sobre
tratamientos consensuados.
La
primera batalla a la que se enfrentan los pacientes y sus familias es la de
obtener un diagnóstico: es a menudo la lucha más desesperante. Esta batalla se
repite a cada nueva etapa de una enfermedad rara degenerativa o en evolución.
La falta de conocimiento de su rara patología pone a menudo en riesgo la vida
de los pacientes y da como resultado una enorme pérdida: retrasos inútiles,
consultas médicas múltiples y prescripciones de medicamentos y tratamientos que
son inadecuados o incluso perjudiciales. Porque se conoce tan poco sobre la
mayoría de las enfermedades raras, un diagnóstico preciso normalmente se hace
tarde, cuando el paciente ya ha sido tratado – durante muchos meses o incluso
años- de otra enfermedad más común. Con frecuencia, solo se reconocen y tratan
algunos de los síntomas.
El paciente de una enfermedad rara es el huérfano de los
sistemas de salud, a menudo sin diagnosis, sin tratamiento, sin investigación y
por consiguiente sin razón para la esperanza.
Bibliografia.
Facultad de Mecidina, Universidad Francisco Marroquin / Sindrome de Proteus / Guatemala, Guatemala / 2008. Citado el 30 de Mayo de 2012. Disponible en: http://medicina.ufm.edu/cms/es/Sindrome-de-Proteus